
How to ensure Design Validation for a Medical Device?
Design Validation Medical Device / Design Verification / V-Cycle / ISO 14971 / ISO 62366 / ISO 10993 / ISO 60601-1 and 60601-1-2 / IEC 62304 / 21 CFR 820.30 / Design History File
Design Validation Medical Device is a critical process encompassing Design Validation, Design Verification, and the V-Cycle methodology.
ISO 14971, ISO 62366, ISO 10993, ISO 60601-1, 60601-1-2, and IEC 62304 standards guide this comprehensive journey within the Design History File. These files are also necessary for CE marking and for the marketing a product in the USA and EU.
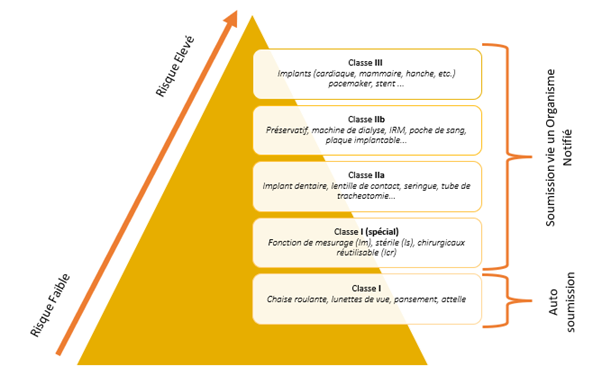
Unlike the Validation of laboratory equipment or software, medical devices require more than a qualification file. It’s imperative to understand the device and create a file that follows the V-cycle. This entails user and functional requirements, risk analysis, validation plans, and Installation, Operational, and Performance Qualification protocols and reports. Classification (I, IIa, IIb, III) significantly shapes this process.
The V-cycle process ensures that the product meets both regulatory requirements and user expectations, by demonstrating the safety of the product and its performance.
Design History File
The validation of a medical device is an integral part of what is called the Design History File. This file encompasses all the documentation of a medical device from the feasibility studies through its development, to the verification, validation and transfer to production activities.
Design Verification phase and Design Validation phase
After completing and validating the phases of “Design and Development Planning,” “Design Input,” and “Design Output,” the project team initiates the “Design Verification” phase for the new medical device’s development. Subsequently, this is followed by the “Design Validation” phase.
These two phases can be associated with the classical concept of Qualification / Validation, applicable to equipment, software, processes, and more. However, within the realm of medical devices, we transcend the mere verification of proper product functionality. Instead, we engage in the verification and validation of all aspects pertaining to the development of the medical device.
The regulations and in particular the new MDR regulations, like the 21 CFR 820.30, require several points in the context of the verification and validation of the medical device, of course some points are to be modulated according to the type and class of the medical device. ISO standards guide us in these different actions.
Different aspects to verify in Design Validation Medical Device
Let’s take a look at these different points to ensure proper verification and validation!
Biocompatibility
Verify the biocompatibility of the medical device (except if the medical device is software only) via test protocols and reports on the characterization of the different materials used in the design of the medical device and via biocompatibility studies (nature and duration of contact between the different materials and the human body or a substance which will then be in contact with the human body). We can refer to the ISO 10993 standard series.
Electro-medical devices
Check the electrical safety and electromagnetic compatibility, if it is an electro-medical device. It is verified that the electro-medical device does not induce, in particular, any unacceptable electrical risk in normal conditions of use or in first fault condition, the test conditions are the least favorable configuration for the product. For the verse on electromagnetic compatibility, it is a question of verifying the immunity of the device to electromagnetic radiation but also verifying its electromagnetic emissions in order to limit disturbances in relation to other medical equipment and devices present in the same environment.
We can refer to the ISO 60601-1 and 60601-1-2 standards.
Software verification
Verify the software if the medical device is software or has embedded software. Depending on the safety class of the software defined by the IEC 62304 standard (A, B or C), the software verification effort and the type of tests required will not be the same. The verification demonstrates that the software functions effectively and meets the issued requirements (software system verification). Furthermore, for classes B and C, it necessitates unit verification and integration verification tests.
Shelf life
Checking the shelf life in its packaging (and thus its stability) before use are also points to be verified within the framework of the Validation for medical devices.
Overall design verification
Verify the overall design (including the instructions for use and labels) of the medical device, whether it meets the stated needs and requirements, whether its performance is as expected, … Also verify the claimed lifetime of the medical device and validate its suitability for use in order to prevent hazards related to possible misuse.
Sterile Packaging
Validate the packaging chosen and the transport conditions, particularly for sterile medical devices where you must preserve the integrity of the sterile barrier.
Sterilization
Validate the sterilization of the medical device (if necessary) and the methods used to ensure its complete sterilization. You must validate the detailed methods for the end user to clean and/or disinfect the medical device to prove their effectiveness.
MRI compatibility
Check the MRI compatibility of the medical device if during its life it may be in contact with or in close proximity to an MRI.
Conclusion
In conclusion, ensuring the validation of a medical device is a meticulous and multifaceted process that transcends the conventional boundaries of qualification and validation. This intricate journey is guided by the principles of the V-cycle, a framework that encapsulates the user requirements, functional specifications, risk evaluations, and comprehensive validation protocols. The classification of the medical device itself, be it I, IIa, IIb, or III, significantly influences the validation strategy.
Integral to the Design History File, this validation endeavor encompasses the entire lifecycle of a medical device, from conception and development through verification, validation, and eventual production. It is an amalgamation of numerous components, each meticulously scrutinized to ensure patient safety, regulatory compliance, and optimal functionality.
The evolving landscape of medical device regulations, especially with the advent of MDR regulations, necessitates adherence to a stringent set of verification and validation criteria. ISO standards provide crucial guidance, offering a roadmap to navigate the intricacies of this validation odyssey.
From biocompatibility assessments to software validation, from electrical safety checks to sterile packaging validations, the holistic approach to medical device validation leaves no stone unturned. It encompasses diverse aspects like electromagnetic compatibility, software veracity, shelf life verification, and MRI compatibility, each playing a vital role in the device’s overall efficacy and patient well-being.
As the medical device industry continues to evolve and regulations grow more stringent, the pursuit of validation becomes more than just a procedural formality. It is the embodiment of an industry’s dedication to innovation, safety, and patient-centered excellence. In embracing these challenges and surmounting the intricacies of medical device validation, we pave the way for the advancement of healthcare technologies that redefine the quality of care and the well-being of humanity.
Discover : Ensuring the environmental qualification of new production areas on a former site – Alispharm



